Introduction
The tumor microenvironment (TME) has long been known to play a central role in tumor progression and immune evasion. An established tumor generally resides in a complex microenvironment (Fig. 1) which nests different types of cellular components including tumor-associated stromal, vascular, and immune cells, in addition to molecular components like interleukins, ROS, VEGF, etc., that support tumor growth (Fig. 2). The immune cell population may be comprised of multiple cell types, including macrophages, dendritic cells, cytotoxic T-cells, T-helper cells, regulatory T cells (Treg), plasma cells, natural killer (NK) cells, and myeloid derived suppressive cells (MDSCs). The constant interaction between the TME and the tumor cells results in varying levels of tumor immunogenicity during tumor growth, treatment, and post-treatment stages. Infiltration of immune cells in the TME is an indication of host immune surveillance, at least during the early stages of tumor development. But, as the tumor evolves and accumulates additional alterations, shifts in the immune response can occur; anti-tumor immune cells can be replaced by pro-tumorigenic or exhausted immune cells, which no longer attack the tumor cells. Furthermore, the tumor cells can also become “invisible” to functional immune cells by eliminating the presentation of tumor neoantigens on the cell surface. These changes indicate an impairment or loss of host immune surveillance, leading to tumor cell escape from host immune response. Importantly, this shift can also play a major role in therapy resistance (Whiteside T. 2008).
The advent of immunotherapies, especially immune checkpoint inhibitors, has revolutionized cancer treatment. Immune checkpoint inhibitors (ICI) are emerging as a standard of care in multiple cancer types. Although approved biomarkers such as CTLA-4, PD-1/PD-L1, TMB, and microsatellite stability index (MSI) have shown utility in predicting response to ICIs, clinical study results have been mixed (Lu J. 2014, Yun S. 2016). In addition,, a proportion of responders within the treated patient populations developed progressive, refractory tumors at later points (Syn N. 2017). Further, in some clinical studies, response to therapy did not correlate with the status of the biomarkers (Cherkassky L. 2022, Keenan T. 2019). Such wide variations in clinical study outcomes have incited an increasing focus in the TME as a potential source of biomarkers with improved predictive power.
Given the pivotal role of TME in disease progression, extensive research in this space has reported a range of prospective biomarkers of response to ICI and other immunotherapies. Several immune correlates have been identified through comprehensive, immunogenomic profiling of the tumor and its microenvironment, including, tumor neoantigens, mismatch repair deficiency (dMMR)/MSI, immune cell infiltrates, alterations in antigen processing and presenting machinery (APM), and onco-viruses (Keenan T. 2019). It has also been noted that individual biomarkers may be insufficient to accurately predict response to immunotherapies in certain tumor-types and tumor-stages, due in part to the frequent shifts in tumor immunogenicity (Kim H. 2022). Aligned with this observation, a growing number of research data have shown that combination of tumor cell-centric biomarkers with TME-based biomarkers, have improved predictive power compared to tumor cell-centric biomarkers alone (Sankar K. 2022). Therefore, comprehensive profiling of the tumor and the TME, especially in the pre-treatment phase, will be required for the identification of composite biomarkers in selecting patients for precision therapies.
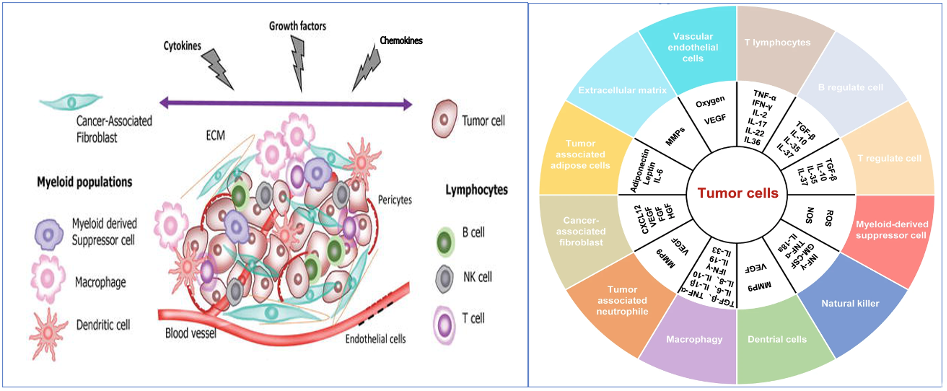
Figure 1 (left): A schematic representation of the complex network of cellular and noncellular components in the TME which facilitates tumor growth and immune evasion. From Alsibai, K. D. , & Meseure, D. (2018). Significance of Tumor Microenvironment Scoring and Immune Biomarkers in Patient Stratification and Cancer Outcomes. In (Ed.), Histopathology – An Update. IntechOpen. https://doi.org/10.5772/intechopen.72648
Figure 2 (right): Cellular and molecular components in the TME From: Jiang X et al. 2020
Major Components of Tumor Milieu
Tumor neoantigen load
Tumors with high tumor mutation burden (h-TMB) have historically been predicted to also have high tumor neoantigen load (h-TNL), and be more likely to elicit response to immunotherapies. On this basis, and supported by clinical studies, h-TMB is an approved biomarker for ICI. However, TMB does not always correlate with TNL (Rizvi N et al. 2015), as not all somatic mutations will generate neoantigens to be presented to CD4+/CD8+ T-cells (Gong L et al. 2021). This limitation may partly explain why only a subset of patients with h-TMB status benefit from ICI treatment. In addition, TMB levels are highly variable across different malignancies (Schumacher T and Schreiber R. 2015, Xu F et al. 2022). Certain cancer types present low TMB, which makes it challenging to identify potential responders to ICI in these cases. To address this gap, many studies have evaluated TNL as a potential biomarker by leveraging various methods of identifying epitopes with a high probability of recognition by cognate T-cells. Results from these studies have expanded the opportunity for immunotherapies in multiple cancer types (Yarchoan M. 2017). Retrospective analysis of tumor samples from clinical studies have reported a significant correlation between h-TNL and clinical benefit in patients treated with ICI, suggesting the importance of neoantigen load in determining response. Results from these studies have demonstrated that neoantigen-directed effector T-cells recognize and kill tumor cells (Rooji et al. 2013, McGranahan et al. 2016, Rizvi N et al. 2015). Further, immunogenomic tumor profiling also revealed a correlation between certain driver mutations and h-TNL. For example, patients with tumors driven by mutations in oncogenes (KRAS, TET1), tumor suppressors (TP53, BRCA1/2), mismatch repair (dMMR) genes, or homologous recombination genes (BRCA1/2), also carried h-TNL, and were associated with durable clinical benefit (Zou X et al. 2021). Currently, immunotherapy based clinical trials are beginning to include TNL as one of the eligibility criteria, or as a study outcome measure to assess its predictive value in certain cancer types. Given the central role of tumor neoantigens in initiating anti-tumor immune response, neoantigen repertoires are also being investigated to identify targets for personalized cancer vaccines, T-cell therapies, and other immunotherapies (Gu Y. 2021).
Advanced sequencing technologies and prediction algorithms have enhanced the ability to identify and prioritize cancer neoantigens for immunotherapy. Neoantigen prediction strategy involves either a direct or an indirect approach. The direct approach is based on analyses of MHC ligandomes eluted from the peptide-MHC complex, using high-throughput mass spectrometry (MS). The direct approach is time consuming and labor intensive, and its accuracy is dependent on the depth of the analysis. Due to these limitations, most studies adopt indirect approaches involving next-generation sequencing and in silico prediction algorithms. The indirect methods start with the identification of tumor specific alterations from tumor/normal whole exome sequencing (T/N WES) (Robbins et al. 2013), or combined T/N WES and tumor RNA sequencing data (Karasaki et al. 2017). Using the sequencing data, candidate neoantigens are identified by applying in silico prediction algorithms like NetMHCpan, which are trained on MHC ligandomes databases derived from in vitro binding affinity assays or MS (Chen I. 2021). SHERPA® is an advanced, pan-allelic MHC-peptide algorithm for predicting antigen binding and presenting, with a high degree of accuracy (Pyke et al. 2021).
Tumor infiltrating lymphocytes
An immune response elicited by tumor neoantigens is reflected by the presence of tumor infiltrating lymphocytes (TIL). Treatment with an ICI can activate infiltrated T-cells by blocking immune checkpoints and inducing tumor regression. Indeed, high TNL was significantly associated with marked infiltration of activated CD4+ and CD8+ T-cells in clinical studies (Sneddon S. 2019). Further, a significant association between TIL, especially CD8+ T cell infiltration, and response to ICI therapy has been observed in treated patients. In a small cell lung cancer (SCLC) cohort, ICI treated patients with PD-L1positive and CD8+ TILhigh density experienced a median PFS that was twice the median PFS observed in patients with PD- L1positive and CD8+ TILlow density tumors (Kanemura H et al. 2022). Recently, a study on an HCC patient cohort from The Cancer Genome Atlas (TCGA) revealed a subset of patients who showed unprecedented response to ICI combination therapy despite low TMB. Analysis of the TME revealed a high CD8+ T cell score in these patients, which was also associated with higher expression of immune checkpoint receptors/molecules and increased tumor infiltrate lymphocytes (TIL). These results suggest that immune inflamed (“hot”) tumors may show better adaptive response to ICI, even when TMB is low (Cherkassky L. 2022). Such studies are paving the path for a deeper investigation of TIL as a biomarker for immunotherapy response prediction.
Treatment induced shift in tumor immune microenvironment
The tumor immune microenvironment can evolve during anti-cancer treatment, leading to adaptive resistance. Patients with “hot tumors” who initially responded to ICI often developed therapy resistance at a later stage. Adaptive resistance can be caused by overexpression of alternate immune checkpoints (Koyama S. 2016), somatic alterations in HLA alleles (Shukla S. 2015), or shifts in tumor immunogenicity initiated by new somatic mutations (Zou X et al. 2021). Therapy resistance has also been associated with loss of tumor neoantigens in some malignancies (Anagnostou V. 2017), or mutations in MHC class I genes like beta-2 microglobulin (Zaretsky J. 2016). In “cold tumors”, for which chemo-/radio- therapy are standard of care, a shift in TME can occur post- treatment. A review of glioblastoma tumor data reported instances of upregulation of immune checkpoint molecules in tumor-infiltrate CD8+ T-cells, leading to T-cell exhaustion (Dai B. 2018). In such cases, response rates to ICI are thought to be influenced by crosstalk between tumor infiltrated CD8+ T-cells and immune checkpoint receptors. These results suggest that, in some cancers, the decision to administer ICI treatment could be informed by immune checkpoints and immune cell infiltration in the TME (Shadbad M et al. 2021).
Antigen processing & presenting machinery (APM)
The APM, a core component of the adaptive immune response against cancer, is functionally driven by several genes. The presence of mutations in one or more APM genes has been correlated with tumor grade, stage, and recurrence. Mutated or absent APM genes can disrupt MHC class I peptide formation and recognition by cognate T-cells. Defective APM components can accelerate tumor cell immune escape, as the ability of cytotoxic T-cells to recognize and kill tumor cells is lost. The impacts of defective APM genes have been documented in both solid and hematologic malignancies. The frequency and types of defects vary across tumor types and include mutations in β2 microglobulin and TAP genes, hypermethylated MHC gene promoter regions, loss-of-heterozygosity (LOH) on chr 6 of the MHC class I gene loci, and others. A few examples of alterations that disrupt MHC class I functioning are shown in Table 1 (Leone P. 2013). Functional loss of MHC class I proteins also leads to reduction of natural killer (NK) cell activation. Indeed, NK cell presence is often low in the TME as tumor cells induce NK cell apoptosis or functionally impair them by binding to CD8+ T-cells (Campoli M and Ferrone S. 2008).
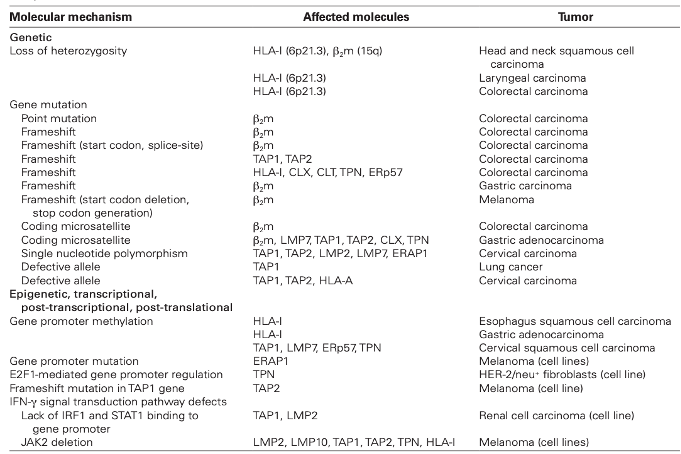
Table 1: Type of alterations observed in MHC class I APM genes (Leone P. 2013).
Microsatellite stability index (MSI)/deficient mismatch repair (dMMR) genes
MSI status is determined based on mutations in one or more of the four MMR genes – MSH2, MSH6, PMS1, and PMS2. Not surprisingly, MSI status has been associated TME changes in some cancers. In colorectal cancer (CRC), patients with MSI high (MSI-H) tumors showed increased efficacy of ICI compared to patients with MSI stable/low (MSS/MSI-L) tumors (Lin A. 2020). Lin A et al., performed a systematic analysis of TME in MSS/MSI-L versus MSI-H status CRC to understand the mechanism behind response to ICI. In the MSI-H tumors, the TME was “hot” with significantly higher number of CD8+ and CD4+ T-cells, T Helper cells, NK cells and M1 macrophages and fewer Tregs (regulatory T-cells). In contrast, in the MSS/MSI-L tumors, the TME was “cold” with significantly upregulated Tregs. Comparison of the gene expression profile (GEP) of immune related genes revealed a significant upregulation of antigen presenting genes, chemokines, cytokines and the tumor necrosis factor receptor (TNFR) family of genes in the MSI-H group. Such differences in the TME suggest that genomic instability can influence tumor immunogenicity.
Tumor angiogenesis in the TME
The TME can induce unregulated and uncontrolled angiogenesis, to support tumor cell growth and metastasis. Tumor associated cells nested in the TME promote tumor angiogenesis by secreting angiogenic factors. The mesenchymal cells, adipocytes and fibroblasts secrete pro-angiogenic growth factors (eg., FGF, VEGF), cytokines (i nterleukins), and chemokines. In addition, non-coding RNAs, like microRNAs (miRNAs), long non-coding RNA (lncRNA), and circular RNAs (circRNAs) in the tumor cells have been found to communicate with the TME to support tumor angiogenesis and metastasis. As tumor cells rely on neo-angiogenesis for nutrition supply, anti-angiogenesis therapies are a part of standard of care in multiple cancers (Jiang X et al. 2020). Multiple genes are involved in tumor specific angiogenesis, and angiogenesis gene signatures are associated with tumor prognosis, TME shift, and response to targeted therapies (Yang Y. 2021).
Oncoviruses in the TME
Certain viral infections are known to induce cancer. Viral factors and host immune response have a complex interaction. Inflammation, which can arise for a range of reasons, can activate cancer-causing viruses that reside in the host tissue. The oncoviruses, in turn, can intensify inflammation leading to tumorigenesis. Oncoviruses can also transform activated immune cells to immunosuppressive cells, further fueling tumor growth. Oncomodulatory viral proteins also stimulate angiogenesis agents, a vital factor in tumor growth (Blaylock R. 2019). Some common oncoviruses include EBV, HCV, HBV, HPV, KSHV, MCV, and HTLV. Analysis of the TME to detect the presence of oncoviruses is critical to inform therapy decisions (Yang Y. 2021).
Future theranostic path for immunotherapy
In clinical settings, the relationship between any single biomarker and response to a given therapy is not expected to be uniform across different cancer types, or even among patients with the same cancer type. This variability in response can arise due to two main reasons: i) differences in the passenger mutations and their number, even if the driver mutations are same across the patient population, and ii) highly divergent HLA alleles at individual patient level (except between identical twins). As a result, the level of tumor immunogenicity which determines response to immunotherapy will be different in each patient. Therefore, even for advanced immunotherapies, the success rate will depend in part on the accuracy with which treatment efficacy is predicted. This translates to characterizing the tumor and its microenvironment for each patient. In scenarios in which a single biomarker is insufficient for response prediction, a combination of complimentary biomarkers may be required (Matsushita H et al. 2016, Zou X et al. 2021). In such cases, comprehensive immunogenomic profiling will be necessary to identify effective, composite biomarkers.
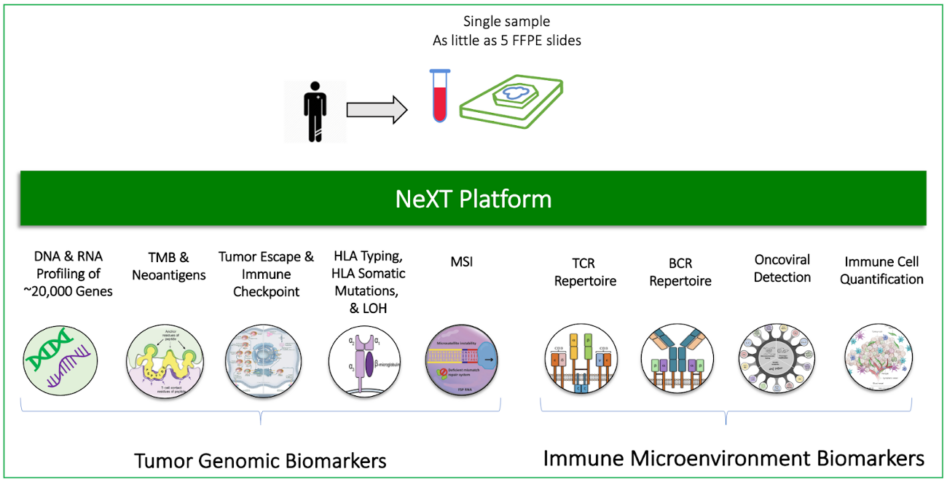
Figure 3 (right): Comprehensive profiling of the tumor and tumor microenvironment with the ImmunoID NeXT Platform®
At Personalis we continue to innovate and address the challenges involved in identifying biomarkers of high predictive power for determining response to targeted and immunotherapy. The ImmunoID NeXT Platform® is designed to provide a comprehensive, multi-dimensional view of the tumor and the tumor microenvironment. It employs a robust assay and integrates highly advanced prediction algorithms to deliver critical information related to tumor drivers and immunogenicity. For example, SHERPA is an advanced, neoantigen prediction algorithm that takes into account the HLA/peptide binding affinity, and further expands by integrating the APM status, gene expression data, and peptide flanking regions to provide a more accurate prediction of tumor neoantigen score. Visibility into driver and passenger alterations, immune landscape, and the potential mechanisms behind immune evasion and therapy resistance not only allows for the identification of composite biomarkers, but also raises the prospect of discovering novel, effective biomarkers to advance precision oncology. ImmunoID NeXT® facilitates all of this using a single assay with least sample quantity, and with it, the capacity to improve clinical outcomes and usher in a new generation of anti-cancer therapies.
To learn more on how ImmunoID NeXT Platform® can help accelerate your translational research efforts, please visit www.personalis.com/immunoid-next-platform/.
All products described here are for Research Use Only and not for use in diagnostic procedures (except as specifically noted).






